
Diagnostika a léčba Pagetovy kostní choroby: aktualizace 2020
Diagnosis and treatment of Paget´s disease of bone: update 2020
Paget’s disease of bone is a progressive focal disorder of bone metabolism characterized by an accelerated rate of bone remodeling, resulting in overgrowth of bone at selected sites and impaired integrity of affected bone. The risk of developing Paget’s disease increases with age and its onset is typically after age 55. Both genetic and environmental causes are thought to contribute to its pathogenesis. The main clinical manifestations are pain, deformities and secondary consequences of bone overgrowth in affected areas, such as osteoarthritis or nerve impingement, although many patients are asymptomatic. Radionuclide bone scan, in addition to targeted radiographs, are recommended as a means of fully and accurately defining the extent of metabolically active disease. In most patients, the serum alkaline phosphatase is elevated. Patients with Paget’s disease who are symptomatic or at risk of complications should be treated with aminobisphosphonates. Zoledronic acid is recommended as first-line therapy producing sustained remissions in almost all patients.
Keywords:
diagnosis – Paget’s disease of bone –treatment – zoledronic acid
Autoři:
Zikán Vít
Působiště autorů:
Osteocentrum, III. interní klinika 1. LF UK a VFN v Praze
Vyšlo v časopise:
Clinical Osteology 2020; 25(4): 189-200
Kategorie:
Doporučené postupy
Souhrn
Pagetova kostní choroba je benigní fokální progresivní porucha kostního metabolizmu charakterizovaná vystupňovanou kostní remodelací, která vede k nadměrnému lokálnímu růstu architektonicky abnormální kosti. Prevalence Pagetovy choroby stoupá s věkem a diagnostikuje se obvykle po 55. roce života. V patogenezi onemocnění se uplatňují genetické a pravděpodobně i environmentální příčiny. Mezi klinické projevy onemocnění patří bolest, deformity a sekundární důsledky nadměrného lokálního růstu kosti, jako je artróza nebo útlak nervových struktur. Většina pacientů ale zůstává bez příznaků. Kostní scintigrafie společně s cíleným rentgenovým vyšetřením je ve většině případů dostačující k určení diagnózy a stanovení rozsahu metabolicky aktivního onemocnění. U většiny pacientů je zvýšená aktivita alkalické fosfatázy v séru. Pacienti, kteří jsou symptomatičtí nebo mají riziko komplikací, by měli být léčeni aminobisfosfonáty. Kyselina zoledronová je lékem první volby, který vede k dosažení trvalé remise u většiny pacientů.
Klíčová slova:
diagnostika – kyselina zoledronová – léčba – Pagetova kostní choroba
Definice a epidemiologie
Pagetova kostní choroba je benigní progresivní metabolické onemocnění skeletu charakterizované lokalizovanou poruchou kostní remodelace s vystupňovanou kostní resorpcí a chaotickou novotvorbou abnormální kostní hmoty, která má primitivní (plsťovitou) strukturu a je více náchylná k deformitám a zlomeninám. Onemocnění postihuje pouze určité místo jedné nebo více kostí (monoostotické nebo polyostotické postižení). Nejčastěji postižené oblasti skeletu zahrnují lebku, páteř, pánev a dlouhé kosti dolních končetin. Chorobu poprvé popsal v roce 1877 anglický chirurg James Paget, který ji nazval osteitis deformans [1]. Pagetova kostní choroba je po osteoporóze druhým nejčastějším onemocněním skeletu, ale její etiologie nebyla dosud objasněna. Odhady prevalence onemocnění se v závislosti na věku pohybují od 1 do 9 % [2,3]. Incidence Pagetovy choroby se přibližně zdvojnásobuje v každé dekádě po 50. roce života [2]. Případy ve věku pod 40 roků patří ke vzácným výjimkám. Onemocnění je o něco častější u můžů než u žen (1,4 : 1) [2] a v určitých etnických skupinách [3]. Mezi země s častým výskytem patří Anglie, Skotsko, země střední Evropy a Řecko, dále Austrálie, Kanada a USA. I v těchto zemích ale existují geografické oblasti s častějším výskytem nemoci [1]. Naproti tomu ve skandinávských zemích a v Asii nebo v Africe je onemocnění hlášeno jen zřídka [5]. Rozpoznání geografických i familiárních výskytů choroby vedlo ke hledání jak environmentálních, tak genetických příčin onemocnění [6,7]. Ukazuje se, že za poslední čtvrtstoletí prevalence Pagetovy kostní choroby klesá (s výjimkou některých regionů) [8,9], rozsah postižení skeletu se zmenšuje a případy symptomatického onemocnění jsou méně časté [9–11].
Histopatologie a patofyziologie
Pagetova kostní choroba je v současné době považována za onemocnění osteoklastů, resp. poruchu regulačních mechanizmů diferenciace, aktivace a přežívání osteoklastů. Osteoklasty u pacientů s Pagetovou chorobou kostí jsou svým vzhledem bizarní, mnohojaderné a nadměrně početné. Zrychlený kostní obrat má za následek abnormální ukládání lamelární kosti proložené neplnohodnotnou (plsťovitou) kostí. Kosti mají neuspořádaný vzhled a variabilně zesílené trabekuly lemované četnými osteoblasty. Neorganizovaná plsťovitá kost zvyšuje objem kosti, což vede k deformitám, zlomeninám a dalším komplikacím. Kost je výrazně vaskularizovaná a kostní dřeň je nahrazována vaskulární fibrózní tkání. Proces může lokálně progredovat nebo se postupně zastavit a změněná kost rekalcifikuje (vzniká sklerotické ložisko). Při polyostotické formě je možné u jednoho pacienta zachytit najednou různá stadia tohoto kostního procesu.
Diferenciace a aktivace osteoklastů vyžaduje signální cestu zahrnující ligand pro receptor aktivující nukleární faktor kB (RANKL) a faktor stimulující kolonie makrofágů (M-CSF). Osteoklasty z postiženého úseku skeletu vykazují zvýšenou citlivost na kalcitriol a RANKL. V jádrech patologických osteoklastů lze zaznamenat intranukleární inkluzní tělíska, ale nikoli v jádrech osteoblastů nebo v osteoklastech z nedotčené kosti u stejného pacienta. Je zatím záhadou, proč jsou postiženy pouze některé oblasti skeletu (nebo jen část určité kosti). Antiresorpční léčba aminobisfosfonáty potlačuje osteoklastickou aktivitu i patologických osteoklastů a obnovuje remodelaci kostí na normální úroveň a u mnoha pacientů vede k dlouhodobé klinické remisi onemocnění. Předpokládá se, že v patogenezi onemocnění se uplatňuji jak genetické, tak environmentální příčiny [12]. Onemocnění se vyskytuje v rodinné i sporadické formě. Pozitivní rodinná anamnéza byla dokumentována u 12 až 40 % pacientů [13,14]. Pacienti s familiární Pagetovou chorobou mají většinou dřívější nástup onemocnění a větší rozsah postižení skeletu ve srovnání se sporadickými případy onemocnění [14–16]. Dědičnost familiární Pagetovy kostní choroby je autosomálně dominantní s variabilní penetrancí [17]. Celogenomové asociační studie a analýzy kandidátních genů identifikovaly celkem 15 genových lokusů, které jsou spojeny s Pagetovou kostní chorobou [18–22]. Většina z těchto rizikových lokusů kóduje proteiny, o nichž je známo, že přímo ovlivňují aktivitu signální dráhy RANK-RANKL, například lokus TNFRSF11A, který kóduje RANK. První a nejlépe dokumentovaná mutace byla nalezena v doméně spojené s ubikvitinem SQSTM1, která kóduje sekvestozom 1. Studium rodinných případů z více zemí vedlo k identifikaci 28 různých mutací SQSTM1 spojených s touto chorobou, z nichž mutace P392L je zdaleka nejběžnější, vyskytující se až u 50 % familiárních případů Pagetovy kostní choroby [23,24]. Přítomnost mutací SQSTM1 koreluje se závažnějším klinickým fenotypem [25]. Avšak u dospělých s mutacemi SQSTM1 zděděných od rodičů s Pagetovou chorobou bylo onemocnění průkazné pouze u 17 % a rozsah postižení byl mírnější ve srovnání s nemocným rodičem [26]. To zdůrazňuje význam interakcí gen-prostředí pro rozvoj onemocnění. Mnoho mutací SQSTM1 zvyšuje aktivaci NFkB, a tím diferenciaci osteoklastů [27], zatímco jiné mutace spojené s Pagetovou kostní chorobou mohou ovlivňovat mikroprostředí kostní dřeně [28]. Ani myši transgenní pro lidský P392L SQSTM1, ani myši s ekvivalentní mutací na endogenním lokusu neonemocní Pagetovou kostní chorobou, i když jejich osteoklasty mají některé vlastnosti patologických osteoklastů (např. zvýšená citlivost na RANKL a TNF) [29]. Byly popsány 3 vzácné autosomálně dominantní skeletální dysplazie, které sdílejí některé rysy s Pagetovou kostní chorobou (zvětšené osteoklasty, nárůst objemu kosti a deformity), ale mají další projevy, které je činí klinicky odlišnými, včetně věku nástupu, ztráty sluchu a časné ztráty dospělých zubů. Pagetova kostní choroba s časným začátkem, familiární expanzivní osteolýza a expanzivní skeletální hyperfosfatázie jsou všechny způsobeny mutacemi v genu TNFRSF11A kódující RANKL a vedou ke zvýšené aktivitě osteoklastů. Čtvrtá porucha, nazývaná juvenilní Pagetova choroba, je autosomálně recesivní kostní dysplazie s nástupem v dětství, která je nejčastěji způsobena zárodečnou mutací genu pro osteoprotegerin, což vede k jeho deficitu a nadměrné aktivaci osteoresorpce.
Ve srovnání s genetickými nálezy jsou důkazy týkající se potenciálních vlivů prostředí, včetně možné role virové infekce, limitované. Nejsou zatím známy žádné modifikovatelné faktory, které přispívají k patogenezi Pagetovy kostní choroby. Byla zkoumána zejména souvislost s virem spalniček a předpokládá se, že očkování proti spalničkám vysvětluje klesající výskyt choroby v některých zemích. Počáteční pozorování poukazovala na intranukleární inkluze v osteoklastech postižených úseků skeletu připomínajících paramyxoviry se strukturou podobnou nukleokapsidům spalniček. Exprese nukleokapsidového proteinu viru spalniček (MVNP) v normálních myších osteoklastech podporovala fenotyp patologických osteoklastů (podobný osteoklastům z místa léze Pagetovy choroby) [30,31] a způsobovala fenotyp choroby v přítomnosti mutace pro SQSTM1 [32]. Exprese MVNP v osteoklastech byla také spojena se zvýšenou diferenciací osteoblastů [33]. Imunohistochemické a molekulární přístupy k potvrzení přítomnosti viru v kostních biopsiích u pacientů s Pagetovou kostní chorobou ale přinesly protichůdná zjištění [34–36]. Možnost, že virová infekce má významnější roli v geneticky citlivém hostiteli, zůstává kontroverzní a je nadále zkoumána.
Klinické projevy
Pagetova kostní choroba se nejčastěji diagnostikuje po 55. roce života. Většina pacientů je asymptomatická a onemocnění je zjištěno náhodně při rentgenovém vyšetření nebo při záchytu zvýšené aktivity celkové alkalické fosfatázy (ALP). Klinické příznaky vykazují značnou variabilitu a závisí na lokalizaci, počtu postižených úseků skeletu, přilehlých strukturách a na metabolické aktivitě choroby. Polyostotická forma se vyskytuje přibližně v 66 %. Postiženy jsou nejčastěji kosti lbi, torakolumbální páteř, pánev, femur a tibie. Méně často jsou postiženy kosti horní končetiny, stejně jako klíček, lopatka, žebra nebo obličejový skelet. Krátké rourovité kosti končetin neonemocní prakticky nikdy. Může docházet k lokální progresi a zvětšení ložiska nebo postupu léze v zasažené kosti. Nikdy ale nevznikají nová ložiska a kost je mimo postižené úseky normální na rozdíl od podobných (Paget-like) vzácných dědičných onemocnění, jako je familiární expanzivní osteolýza, idiopatická hyperfosfatázie nebo juvenilní Pagetova kostní choroba. Nejčastějším příznakem bývá bolest postiženého místa skeletu (někdy nabývá větší intenzity v noci). Etiologie bolesti spojené se změnami v kosti je nejasná. Může to být způsobeno periosteálními změnami způsobenými zvětšením kosti ve spojení s hyperemií nebo mikrofrakturami. Může být vyšší teplota kůže v důsledku zvýšeného prokrvení v místě postižení. Z praktického hlediska je užitečné hodnotit možné symptomy a komplikace z hlediska lokalizace onemocnění. Poměrně často jsou postiženy kosti lebky. Bývá postižen lební kryt a nejednou i spodina. Postižení lebky může vést k deformitám, bolestem hlavy, závratím a vzácně k neurologickým následkům včetně bazilární invaginace, hydrocefalu s nestabilitou chůze, demencí nebo apatií v důsledku zvýšené vaskularity kostní léze a steal (zlodějského) fenoménu [37]. Hluchota se popisuje přibližně u 2 % pacientů (v důsledku postižení spánkové kosti a kochleárního aparátu). Méně často dochází k postižení jiných hlavových nervů (II., V., VI. a VII.). Postižení dolní čelisti nebo maxilárních kostí může mít za následek deformaci čelistí, malokluzi a parodontální onemocnění [38]. Cévám podobné (angioidní) pruhy lze nalézt při oftalmologickém vyšetření až u 20 % pacientů s aktivní Pagetovou kostní chorobou, zejména s postižením lebky [39]. Běžně se vyskytují široké rozšířené žíly na pokožce hlavy. Páteř je zpravidla postižena v hrudní nebo lumbální oblasti. Komplikací mohou být kompresivní deformity obratlových těl a kyfotizace páteře, případně může dojít k útlaku míšních kořenů a k rozvoji motorického deficitu nebo radikulární bolesti. Vzácně dochází ke stlačení míchy nebo její dysfunkci v důsledku ischemie při steal fenoménu [40] a při postižení hrudních obratlů byla dokumentována kvadruparéza nebo paraparéza v závislosti na výši postižení. V oblasti pánve je Pagetova choroba často asymptomatická, ale pokud je v blízkosti kloubu, může vést k bolestem v důsledku rozvoje sekundární artrózy nebo při protruzi acetabula. Dlouhé kosti končetin jsou často postiženy jednostranně. Postižena je hlavně diafýza, která se někdy zakřivuje. Deformity kostí mohou mít za následek změny chůze a zvýšené mechanické namáhání, které vede k bolesti zad a kloubů a zvyšuje rychlost progrese artrózy s nutností náhrady kyčelního nebo kolenního kloubu. Zlomenina, nejčastěji diafýzy femuru nebo v subtrochanterické oblasti, je méně častou, ale závažnou komplikací. Mnohem častější jsou štěrbinovité fraktury na konvexitě deformované dlouhé kosti [41,42]. Zlomeniny u aktivní Pagetovy kostní choroby jsou zpravidla spojeny s větší ztrátou krve. Polyostotické aktivní onemocnění může vést velmi vzácně k dalším komplikacím, např. k srdečnímu selhání (v důsledku hyperkinetické cirkulace a vysokého srdečního výdeje). Vzácně vzniká hyperkalciurie s rizikem urolitiázy nebo v případě imobilizace i hyperkalcemie. Pacienti mají zvýšené riziko primárních kostních novotvarů [2]. Osteosarkom se může objevit v místě postižené kosti a projevuje se zvýšenou bolestí, která špatně reaguje na léčbu, lokálním otokem a méně často patologickou zlomeninou [43–45]. Odhady prevalence osteosarkomu u pacientů s Pagetovou chorobou se pohybují od 0,2 do 1 %. Méně často se objevují obrovskobuněčné nádory, které jsou obvykle benigní [46] a mají několik odlišných charakteristik, včetně predilekční lokalizace (vznikají zejména v oblasti lebky, kostí obličeje a pánve) a pozdějšího věku výskytu nádoru [47].
Biochemické vyšetření
Biochemicky prokazujeme především zvýšení kostní frakce alkalické fosfatázy (bALP) podmíněné zvýšeným počtem osteoblastů, nebo zvýšení markeru syntézy kolagenu propeptidu prokolagenu I v séru (PINP). Jiný marker kostní novotvorby, osteoblasty produkovaný osteokalcin, je však u pacientů s Pagetovou kostní chorobou nepřiměřeně nízký. Záchyt zvýšené katalytické aktivity celkové ALP v séru při normální aktivitě glutamyl transferázy (GGT) může být prvním nálezem, který povede k dalšímu vyšetřování a diagnóze často asymptomatické Pagetovy kostní choroby. Stupeň elevace ALP nebo jiného kostního markeru obecně odráží rozsah a aktivitu onemocnění, ale tato korelace není zcela konzistentní [48–51]. Normální nebo minimálně zvýšená aktivita ALP může být pozorována u pacientů s monostotickým onemocněním a u některých pacientů s polyostotickým onemocněním, např. normální ALP se vyskytuje u pacientů s izolovanou pánevní lézí, s několika postiženými těly obratlů nebo v případě převážně sklerotického postižení kostí. Naproti tomu izolovaná Pagetova choroba lebky může způsobit velmi výrazné zvýšení aktivity ALP [52]. O primárně zvýšeném počtu osteoklastů a zvýšené osteoresorpci svědčí zvýšené koncentrace osteoklastické (tartarát rezistentní) kyselé fosfatázy (TRAP izoenzym 5b) v séru nebo markery degradace kostního kolagenu karboxy nebo aminoterminální telopeptidy kolagenu I (CTX nebo NTX). Je dobré mít na paměti, že biochemické markery vždy vypovídají o celotělové aktivitě kostní remodelace, a nejsou tedy specifické pro určité místo skeletu – na rozdíl od rentgenového nebo scintigrafického vyšetření.
I při značně vystupňované kostní remodelaci nedochází ke změnám metabolizmu minerálů a zjišťujeme normální koncentrace vápníku a fosforu v séru a v moči a normální koncentrace parathormonu. Vzácnější výjimkou je vznik hyperkalciurie nebo hyperkalcemie, např. v případě imobilizace u pacienta s aktivní polyostotickou formou choroby [9]. Naproti tomu hyperkalcemie u ambulantního mobilního pacienta naznačuje přítomnost jiné poruchy, jako je např. primární hyperparatyreóza. Biochemické vyšetření podává informaci o aktivitě choroby a umožňuje též sledování úspěšnosti léčby a průběhu choroby (viz kapitola Monitorování).
Rentgenové vyšetření skeletu
Rentgenové vyšetření, které hodnotí změny struktury postižených úseků skeletu, je pro diagnostiku Pagetovy kostní choroby rozhodující. Rentgenový obraz závisí na stadiu choroby a může někdy činit diferenciálně diagnostické obtíže. Rozlišují se 3 základní stadia choroby: 1. stadium osteolýzy; 2. intermediární (smíšené) stadium osteolýzy s neuspořádanou kostní novotvorbou (hyperostóza) a 3. sklerotické stadium (hyperostotická skleróza). U jednoho pacienta se mohou současně vyskytovat různá stadia choroby. Záchyt čistě osteolytického stadia je vzácný a může vytvářet obraz rentgenologicky neodlišitelný od metastázy nebo lymfomu. Mezi charakteristické rentgenologické nálezy patří [53]: Na lebce lze v časné fázi onemocnění pozorovat mapovitá osteolytická ložiska „osteoporosis circumscripta“. Smíšené lytické a sklerotické léze s postupnou lokální progresí mají za následek zesílení kortikální kosti, ztluštění kostních trámců, nejasné rozlišení kortikální a trámčité kosti a nadměrný růst postižené kosti (obr. 1). Mohou vznikat deformity kosti až mnohočetné štěrbinové fraktury v konvexitě dlouhých kostí [53]. V průběhu času se jednotlivé kostní léze vyvíjejí, jak je uvedeno výše, a může docházet k jejich zvětšení. Postižení pánve, které může mít za následek protruzi acetabula vede k „trojúhelníkovému “vzhledu pánve. Vzhled postižených obratlů u některých pacientů nabývá „slonovinového “vzhledu nebo tzv. „rámového“ obratle (obratel je zvětšený ve všech třech rovinách). Zvětšení obratlů může navíc způsobit ztrátu normální bederní lordózy a zvýraznit dorzální kyfózu.
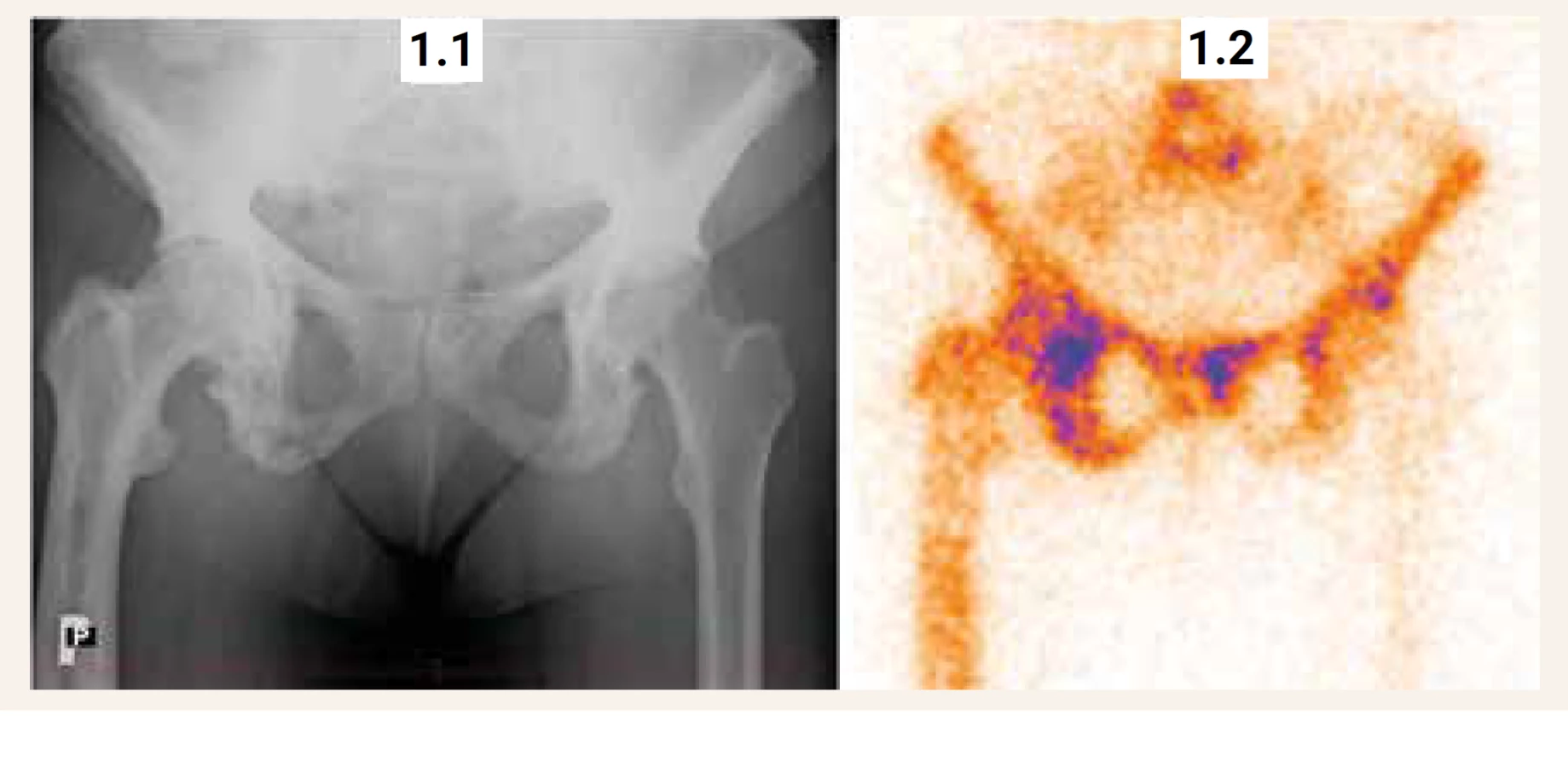
Scintigrafické vyšetření skeletu
Scintigrafie skeletu s použitím 99mTc bisfosfonátu zjistí rozsah metabolicky aktivního postižení skeletu (obr. 1, obr. 2). Zvýšená akumulace radiofarmaka v místech se zvýšenou osteoblastickou aktivitou (a vyšším krevním průtokem) může předcházet typické rentgenové změny, zejména u časných fází onemocnění. V případě monoostotického postižení bývá scintigrafie citlivější metodou v hodnocení aktivity kostního procesu než vyšetření aktivity ALP nebo jiného biochemického markeru kostní remodelace (vypovídá o celotělovém kostním obratu). Také v případě lokalizované bolesti v blízkosti kloubu rozliší scintigrafie, zda aktivita vychází spíše z postižené kosti, nebo z oblasti artrotického kloubu. V případě sklerotického stadia choroby s nízkou metabolickou aktivitou (nebo při efektivní léčbě) může scintigrafické vyšetření vykazovat i negativní nález. Progrese choroby se vždy týká již postižených úseků skeletu. Nálezy na kostních skenech však nejsou specifické a pro potvrzení diagnózy je třeba získat rentgenové zobrazení postižených úseků skeletu.

Diagnostika a diferenciální diagnostika
Diagnostika onemocnění by měla zahrnovat pečlivé anamnestické vyšetření i s ohledem na familiární výskyt onemocnění. Fyzikální vyšetření může odhalit deformity nebo známky artrózy přilehlých kloubů, zvýšenou teplotu nebo napětí kůže nad aktivním ložiskem choroby nebo změny kožní citlivosti a obrny nervů v případě úžinových syndromů. Součástí vyšetření je rovněž vyšetření sluchu, zejména v případech postižení spánkové kosti. Diagnóza Pagetovy choroby kostí je primárně radiologická (viz radiologický nález). Rentgenové snímky postižených míst skeletu jsou nezbytné pro identifikaci muskuloskeletálních důsledků Pagetovy choroby, včetně zlomenin, potenciálních maligních lézí, sekundární artrózy nebo jiných kostních abnormalit. Postup vyšetření u pacienta s podezřením na Pagetovu kostní chorobu znázorňuje schéma. Stanovení aktivity ALP je užitečné pro hodnocení a sledování aktivity onemocnění [48–51]. U pacientů s onemocněním jater nebo charakteristickou kostní lézí, kteří však mají normální aktivitu celkové ALP, je vhodné stanovit kostní specifickou ALP, C-telopeptid (CTx) nebo N-terminální propeptid prokolagenu typu I (PINP) v séru. Tento přístup je obecně v souladu s publikovanými doporučenými postupy [54,55]. Pagetova kostní choroba se vyskytuje u stárnoucí populace, která má zvýšenou prevalenci i dalších onemocnění, např. artrózy a osteoporózy, a tak se symptomatika může v řadě případů překrývat. Diferenciálně diagnosticky je nutné vyloučit osteomalacii, hyperparatyreózu, metastatické postižení skeletu nebo fibrózní dysplazii. U pacientů se zvýšenou aktivitou ALP by měly být stanoveny vždy rovněž sérové hladiny vápníku, fosforu a 25-hydroxyvitaminu D, aby se vyloučily další příčiny tohoto zvýšení, a to vždy před zahájením léčby aminobisfosfonáty (vyloučení osteomalacie, deficitu vitaminu D nebo hyperparatyreózy). Bolesti kostí a zvýšená aktivita ALP může být i u dalších kostních procesů, např. u osteomalacie, u níž ale chybí radiologické nálezy charakteristické pro Pagetovu chorobu, a charakteristický bývá laboratorní nález (hypokalcemie nebo hypofosfatemie). Pseudofraktury (Looserovy zóny) se vyskytují u osteomalacie na konkávní části dlouhých kostí, na rozdíl od Pagetovy kostní choroby, při níž se vícečetné štěrbinové zlomeniny vyskytují na konvexitě kosti. Smíšený osteolytický a sklerotický vzhled Pagetovy kostní choroby může u některých pacientů ztížit rentgenologické rozlišení nádorového procesu. Nové léze na kostním skenu se však u Pagetovy choroby obecně nevyskytují. Jiné zobrazovací technologie, jako je CT nebo MRI, mohou být užitečné při hodnocení kostních lézí, např. při podezření na malignitu [56,57]. CT-vyšetření je přínosné pro ověření, zda je kortikální kost neporušená, a pro stanovení následků zlomenin nebo nadměrného růstu kosti v oblasti páteře nebo lebky (obvykle v těchto lokalizacích nelze rozlišit na prostém RTG-snímku, obr. 2). Vyšetření MRI může pomoci při rozlišení lytických nebo intermediárních lézí Pagetovy choroby od sarkomu [58]. U osteolytického ložiska Pagetovy kostní choroby je na rozdíl od metastatického postižení zachován MRI signál tukové kostní dřeně v T1-vážených obrazech. U části pacientů, kteří jsou dlouhodobě imobilizováni pro frakturu, se může také dramaticky zvýšit osteolýza v postiženém úseku skeletu a nález může být snadno zaměněn za akutní osteomyelitidu nebo maligní proces. Sklerotické stadium Pagetovy kostní choroby může být obtížně odlišitelné od metastatického postižení, zvláště u pacientů se známým karcinomem prostaty nebo prsní žlázy. V tomto případě nebude přínosné vyšetření MRI, jelikož MRI signál ze sklerotického ložiska může být obdobný jako v případě metastázy a může být nápomocné vyšetření pozitronovou emisní tomografií (PET), při němž sklerotické ložisko Pagetovy choroby nevykazuje zvýšenou metabolickou aktivitu. Na druhé straně ale PET/CT může být „falešně pozitivní“ se zvýšenou absorpcí pozorovanou u aktivních kostních lézí při Pagetově kostní chorobě [59]. Pokud zobrazovací vyšetření nejsou schopna tyto procesy dostatečně spolehlivě odlišit, je nezbytné provedení kostní biopsie. Osteosarkom související s Pagetovou kostní chorobou se obvykle projevuje zhoršením lokalizované bolesti kosti a může být doprovázen otokem měkkých tkání v postižené oblasti. MRI může pomoci definovat anatomii měkkých tkání a další změny u malignity. Obrovskobuněčné kostní nádory jsou benigní, ale někdy mohou mít agresivní povahu a projevují se lokalizovanou bolestí, otoky a omezením pohybu kloubů. Tyto nádory se typicky vyskytují v oblasti kostí lebky nebo pánve u pacientů s polyostotickým postižením. Velmi vzácně mohou vznikat i extraskeletální osteoklastomy.

Léčba
Léčba je indikována u pacientů se symptomatickou Pagetovou kostní chorobou a u vybraných asymptomatických pacientů s vysokou aktivitou onemocnění (vysoká aktivita ALP, pozitivní kostní scintigrafie), zejména v případě zvýšeného rizika komplikací u neléčeného onemocnění [60]. Rizikové jsou zejména aktivní léze v oblasti páteře a nosných kostí (riziko patologické zlomeniny nebo útlaku nervových struktur), kostí lebky (riziko ztráty sluchu) nebo kostí, které přiléhají ke kloubu (riziko sekundární artrózy). Cílem léčby je zmírnit bolest a snížit rychlost remodelace kosti, což umožní tvorbu normální lamelární kosti, sníží vaskularizaci kosti a zpomalí progresi onemocnění. Mezi příznaky a nálezy, které mohou reagovat na farmakologickou léčbu u pacientů s aktivním onemocněním, patří bolest kostí (i sekundární v případě komprese nervových struktur), stejně jako rentgenové nálezy osteolytických kostních ložisek. Zlomenina v důsledku abnormální kostní struktury je dalším projevem Pagetovy choroby, při které může být léčba prospěšná. U pacientů s aktivním onemocněním, u kterých je plánován chirurgický výkon v místě léze, např. náhrada kyčelního kloubu, zahajujeme farmakologickou léčbu aminobisfosfonáty alespoň 3–6 měsíců před plánovaným výkonem.
Léčba aminobisfosfonáty
Léčba aminobisfosfonáty 1. a 2. generace v léčbě Pagetovy kostní choroby byla první volbou před zavedením kyseliny zoledronové do klinické praxe, která je významně účinnější při dlouhodobé léčbě. Léčba perorálně podávanými aminobisfosfonáty upravuje při vysokém dávkování kostní obrat a zlepšuje i histologický nález, ale účinky nejsou úplné a dlouhodobé. Z perorálních léků je v současné době dostupný alendronát a risedronát. Alendronát je perorálně podávaný aminobisfosfonát používaný při Pagetově chorobě v dávce 40 mg denně po dobu 6 měsíců [61–63]. Ve dvou studiích byla prokázána účinnost při normalizaci ALP v séru u 63 % a 86 % pacientů léčených alendronátem [62,63]. Risedronát se pro léčbu Pagetovy kostní choroby podává v dávce 30 mg 1krát denně po dobu 2 měsíců [64]. Pacienti, jejichž aktivita ALP nedosahuje normálních hodnot, by měli být léčeni opakovaně. Při vysokém dávkování se ale mnohem častěji než při léčbě osteoporózy vyskytují nežádoucí účinky. Limitujícím faktorem u p.o. bisfosfonátů je především tolerance gastrointestinálního traktu (riziko gastroezofageální iritace nebo zánětu či rozvoje ulcerace).
Kyselina zoledronová je v současné době lékem první volby [60, 65]. U Pagetovy kostní choroby byla po jedné dávce kyseliny zoledronové (5 mg v infuzi) indukována biochemická remise u 96 % pacientů [66]. Remise je po jednorázové infuzi udržována u většiny pacientů od 5 do 6 let [67] a u některých až 10 let; pouze u 14 % pacientů došlo k biochemickému relapsu do 9 let [68]. Kyselina zoledronová je navíc účinná u pacientů dříve léčených jinými bisfosfonáty [69]. Kyselina zoledronová (jednorázová infuze v dávce 5 mg) byla srovnávána s perorálním risedronátem (30 mg/den po dobu 60 dnů) v multicentrické randomizované (dvojitě zaslepené) studii která zahrnula celkem 357 pacientů s Pagetovou kostní chorobou [66]. U obou skupin došlo ke zmírnění bolestí kostí a ke zlepšení kvality života; klinicky významné zlepšení bolestí bylo častější po 6 měsících léčby u pacientů, kteří dostali kyselinu zoledronovou (50 % vs 37 %). Kyselina zoledronová vedla k častějšímu dosažení primárního cílového endpointu: normalizaci ALP v séru nebo snížení rozdílu mezi počáteční aktivitou ALP a průměrnou normální hodnotou o nejméně 75 % (96 % vs 74 %) a byla spojena s kratším mediánem času do terapeutické odpovědi (64 dnů vs 89 dnů) a s mnohem nižší frekvencí ztráty odpovědi v následném sledování po mediánu 190 dnů (1 % vs 27 %). Z 296 pacientů z původní studie si udržovali normální aktivitu ALP v séru ti pacienti, kteří dostávali kyselinu zoledronovou, zatímco u pacientů dříve léčených risedronátem došlo k pomalému, lineárnímu zvýšení ALP v séru během 6 měsíců po ukončení léčby [70]. Během dlouhodobého sledování (6,5 roku) bez další léčby aminobisfosfonáty byl relaps zjištěn jen u 1 ze 152 pacientů, kteří dostali kyselinu zoledronovou [67].
Po aplikaci kyseliny zoledronové se přibližně 2krát častěji než při léčbě risedronátem zjišťují příznaky akutní fáze podobné chřipkovému onemocnění (zvýšená teplota nebo horečka, bolesti hlavy, nauzea, bolesti kostí a artralgie), které vymizí většinou do 2–4 dnů. Z možných nežádoucích účinků je nutné upozornit na potencionální renální toxicitu aminobisfosfonátů, zejména kyseliny zoledronové. Bylo dokumentováno, že vyšší dávky kyseliny zoledronové mohou způsobit tubulární nekrózu a akutní renální selhání. Důležitým opatřením je proto dostatečná hydratace před aplikací infuze. Opatrnost je nutná u pacientů, kteří užívají jiné léky, které mohou významně ovlivňovat funkci ledvin, např. aminoglykosidy nebo diuretika. U pacientů s chronickým onemocněním ledvin s glomerulární filtrací < 35 ml/min/1,73 m2 jsou aminobisfosfonáty většinou kontraindikovány. U některých pacientů však může být po konzultaci s nefrologem zváženo snížení dávky bisfosfonátu. Perorální bisfosfonáty pro léčbu osteoporózy prokázaly účinnost i u pacientů s chronickou renální insuficiencí [71,72], avšak bezpečnost a účinnost dávkování u Pagetovy choroby není známa. Pokud je upřednostňováno intravenózní podání, klinické zkušenosti u pacientů s odhadovanou rychlostí glomerulární filtrace < 35 ml/min/1,73 m2 naznačují, že riziko poškození ledvin u kyseliny zoledronové může být nižší při užití pomalejší rychlosti infuze (60 minut) [73]. Je také doporučováno užití nižší dávky kyseliny zoledronové (2–2,5 mg), i když chybí evidence podporující vyšší bezpečnost. Vzhledem k neurčené úrovni rizika pro osteonekrózu čelisti ve spojení s užíváním bisfosfonátů u Pagetovy choroby by měly být plánované invazivní stomatologické výkony, jako jsou extrakce nebo implantáty, provedeny nejméně 3–6 měsíců před zavedením léčby aminobisfosfonáty, kdykoli je to možné.
Před zahájením léčby aminobisfosfonáty i po celou dobu léčby je nezbytné zajistit normální hladinu vápníku a fosforu v séru a dostatečné zásobení vitaminem D. Zejména u pacientů léčených kyselinou zoledronovou je riziko hypokalcemie, která může být u pacientů s deficitem vitaminu D i symptomatická. Zatímco přístupy k suplementaci vitaminem D se mohou lišit, naší praxí je podávat doplňkový vitamin D3 (1 000–2 000 IU denně nebo 7–14 000 IU týdně), pokud je hladina 25OHD < 75 nmol/l. U pacientů, jejichž hladina 25OHD je ≤ 50 nmol/l, předepisujeme v případě plánované léčby aminobisfosfonáty p.o. suplementaci vitaminem D3 v dávce 28 000–35 000 IU týdně po dobu 6–8 týdnů, přičemž je třeba zajistit kontrolu hladiny 25-hydroxyvitaminu D. Pacientům by měl být také podle potřeby podáván doplňkový vápník v závislosti na příjmu potravy, s cílem dosáhnout celkového denního příjmu 1 200 mg elementárního vápníku.
Další možnosti léčby
Pacienti, kteří netolerují nebo nemohou užívat bisfosfonáty, byli v minulosti léčeni převážně lososím kalcitoninem. Kalcitonin může snížit vaskularitu v místě léze a má rychlý nástup účinku, ale ve srovnání s aminobisfosfonáty je dlouhodobě významně méně účinný a v současné době již není běžně dostupný. Denosumab je plně lidská monoklonální protilátka proti RANK ligandu, která se užívá k léčbě osteoporózy. Mechanizmus účinku denosumabu naznačuje, že by u vybraných pacientů mohl být užitečný i u Pagetovy choroby kostí, např. u pacientů s renální nedostatečností, která vylučuje léčbu aminobisfosfonáty. Dosud však není dostatek údajů a klinických zkušeností, které by podpořily jeho užití v léčbě Pagetovy kostní choroby. Publikované důkazy týkající se denosumabu u Pagetovy choroby jsou zatím omezeny na kazuistiky [74]. U pacientů s chronickým onemocněním ledvin byla dokumentována i těžká hypokalcemie, a léčba proto vyžaduje opatrnost [75,76].
Chirurgické a další intervence
Ortopedické – chirurgické možnosti, které lze zvážit u vybraných pacientů s Pagetovou chorobou, zahrnují kloubní náhrady a plastiky, korekční osteotomii u deformací dlouhých kostí, fixaci zlomeniny, dekompresi páteře a resekci kostních nádorů. Mezi hlášené komplikace totální artroplastiky kyčelního kloubu u pacientů s Pagetovou chorobou kyčle patří nadměrná ztráta krve [77] a zvýšené riziko aseptického uvolnění implantátu s nutností revize výkonu. [78]. U plánovaného chirurgického výkonu v místě aktivní kostní léze se proto doporučuje předoperační léčba aminobisfosfonátem 3–6 měsíců před operací, a to jak ke snížení ztráty krve, tak ke snížení rizika revize [79]. Toto odborné doporučení podporují publikované kazuistiky, které dokumentovaly, že uvolňování kyčelních implantátů bylo častější u pacientů s vyšší aktivitou sérové ALP [80]. U pacientů se zlomeninou v místě patologické léze kosti chybí klinické studie a formální doporučení pro vedení léčby. Bisfosfonáty ale jsou doporučovanou volbou, pokud je pacient stabilní a je vyloučen nedostatek vitaminu D, hypokalcemie nebo hypofosfatemie a jsou zajištěna opatření k prevenci hypokalcemie [81].
Monitorování
Pro monitorování léčby Pagetovy kostní choroby v klinické praxi je nejčastěji doporučeno měření aktivity celkové ALP (v případě normální koncentrace GGT) nebo kostního izoenzymu ALP zpočátku většinou v 3- až 6měsíčních intervalech, aby se posoudila počáteční odpověď na léčbu; dále po 6 měsících [82]. Účinnější bisfosfonáty potlačují kostní resorpci během několika dní až týdnů, což dokazuje snížená koncentrace N-telopeptidů v moči nebo C-telopeptidů v séru (beta-CTX). Následně se snižuje i kostní novotvorba (pokles kostních markerů v týdnech až měsících) [60]. Normalizace ALP v séru je spojena s prodlouženým obdobím biochemické remise a histologickým důkazem normálního kostního obratu; na druhé straně je zvýšení sérové ALP konzistentní se zvýšením aktivity nemoci [62,66]. ALP v séru je obecně doporučované měřítko pro monitorování biochemické odpovědi na léčbu bisfosfonáty. U pacienta s Pagetovou kostní chorobou a současným onemocněním jater používáme pro hodnocení aktivity kostního procesu sérové hladiny propeptidu prokolagenu typu 1 (PINP, markeru syntézy kostního kolagenu). U podskupiny pacientů s monostotickým onemocněním a normálními markery kostního obratu může být při sledování odpovědi na léčbu po 6 až 12 měsících užitečná kostní scintigrafie, která dokáže detekovat aktivní onemocnění a určit jeho rozsah. Přestože již byla popsána normalizace scintigrafie po terapii [83], v klinické praxi scintigrafická aktivita může přetrvávat, což naznačuje, že opakované zobrazovací studie, včetně scintigrafie kostí, nejsou obecně užitečné pro rutinní sledování odpovědi na léčbu. Pokud symptomatika (např. bolest kostí) nereaguje podle očekávání na léčbu, tak je vhodné zvažovat doplnění CT nebo MRI-vyšetření k lepší charakterizaci kostních lézí a vyloučení komplikací onemocnění včetně zlomenin, spinální stenózy nebo stlačení nervů, která se mohou objevit v rámci patologických lézí u Pagetovy kostní choroby nebo neoplazií, jako je osteosarkom, fibrosarkom, chondrosarkom a obrovskobuněčný nádor. U pacientů s normální aktivitou sérové ALP, u kterých byla náhodně zjištěna patologická léze, ale u nichž není léčba považována za vhodnou, pacienta každoročně sledujeme z hlediska bolesti kostí, funkčního poškození a aktivity ALP.
Opakování léčby aminobisfosfonáty: indikace a přístup
Indikace pro opakovanou léčbu bisfosfonáty obecně závisí na symptomatice a evidenci zvýšeného nebo abnormálního kostního obratu (ALP, scintigrafie) nebo rentgenologických známek progrese onemocnění (osteolýza). Nové příznaky, které mohou reagovat na opakovanou léčbu, pokud jsou způsobeny Pagetovou chorobou, by měly být odlišeny od sekundárních nebo nesouvisejících změn, které nejsou způsobeny současnou aktivitou onemocnění. Údaje z klinických studií adekvátně neřeší problém opakované léčby, ale byl navržen následující přístup, který je v souladu s dostupnou evidencí [60]: Opakovaná léčba kyselinou zoledronovou je indikována u pacientů, u nichž se ALP v séru nenormalizuje do 12 měsíců nebo následně stoupne nad normální hodnotu. Opakovaná léčba risedronátem je nutná u pacientů, jejichž sérové hladiny ALP nedosahují normálních hodnot do 2 měsíců od počáteční léčby nebo následně stoupnou nad normální hodnoty. Pokud 2 cykly risedronátu nevedou k normalizaci sérové ALP, je doporučeno užití kyseliny zoledronové. Opakovaná léčba alendronátem může být podána po 6 měsících od ukončení předchozí léčby, pokud byly dosaženy normální hodnoty sérové ALP a poté vzrostly nad normální hodnoty, nebo pokud nebylo dosaženo normálních hodnot během počátečního cyklu. Alternativou k opakované léčbě alendronátem je opět přechod na léčbu kyselinu zoledronovou, jejíž aplikace by ale měla být odložena alespoň o 6 měsíců od ukončení léčby alendronátem.
Prognóza
U neléčených pacientů může dojít k lokální progresi onemocnění s rozšířením osteolytických lézí a k rozvoji kostní deformity [84]. Bolest v souvislosti s aktivní kostní lézí obvykle rychle reaguje na terapii aminobisfosfonáty s postupnou normalizací sérové ALP. Při léčbě aminobisfosfonáty, zejména kyselinou zoledronovou, je účinnost léčby vysoká a léčba je dobře tolerována. Většina studií však nebyla navržena tak, aby potvrdila očekávání, že léčba aminobisfosfonáty předejde dlouhodobým komplikacím [84]. V randomizovaných studiích s aminobisfosfonáty u pacientů s Pagetovou chorobou byla účinnost léčby dokumentována snížením bolesti a zlepšením kvality života, snížením aktivity ALP v séru a normalizací dalších markerů kostního obratu (např. PINP) nebo rentgenovými známkami hojení [66].
Závěr
Diagnostika Pagetovy kostní choroby je často opožděná a řada pacientů má již rozvinuté komplikace. Nejčastěji je diagnostikována náhodně u osob starších 50 let na základě zjištěné zvýšené aktivity sérové ALP nebo rentgenového nálezu. Diagnóza Pagetovy kostní choroby u pacientů s klinickými nebo laboratorními nálezy je potvrzena prokázáním charakteristických rentgenových změn. Scintigrafie skeletu ověří rozsah postižení skeletu; CT- nebo MRI-vyšetření je přínosné pro stanovení následků zlomenin nebo nadměrného růstu kosti zejména v oblasti páteře a lebky nebo při podezření na malignitu (schéma). Kauzální léčba této choroby zatím není známa. Kyselina zoledronová je v současné době nejúčinnější možností léčby, která u většiny pacientů navozuje dlouhodobou remisi onemocnění. Léčba je indikována, pokud je zvýšená metabolická aktivita onemocnění jak u symptomatických pacientů, tak u asymptomatických pacientů, pokud je přítomno riziko rozvoje komplikací. Současně je nezbytné zajistit dostatečné zásobení vitamínem D a dostatečný příjem vápníku jako prevenci hypokalcemie a sekundární hyperparatyreózy. Objasnění etiopatogeneze Pagetovy kostní choroby přinese do budoucna jistě i nové možnosti prevence a léčby.
Publikace byla podpořena MZ ČR – RVO VFN64165.
doc. MUDr. Vít Zikán, Ph.D.
www.vfn.cz
Received | Doručeno do redakce | Doručené do redakcie 4. 12. 2020
Accepted | Přijato po recenzi | Prijaté po recenzii 28. 12. 2020
Zdroje
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60: 37–64.9. Dostupné z DOI: <http://dx.doi.org/10.1177/095952877706000105>.
- van Staa TP, Selby P, Leufkens HG et al. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17(3): 465–471. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.2002.17.3.465>.
- Corral‐Gudino L, Borao‐Cengotita‐Bengoa M, Del Pino‐Montes J et al. Epidemiology of Paget’s disease of bone: a systematic review and meta‐analysis of secular changes. Bone 2013; 55(2): 347–352. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bone.2013.04.024>.
- Altman RD, Bloch DA, Hochberg MC et al. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15(3): 461–465. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.2000.15.3.461>.
- Yip KM, Lee YL, Kumta SM et al. The second case of Paget’s disease (osteitis deformans) in a Chinese lady. Singapore Med J 1996; 37(6): 665–667.
- Siris ES, Ottman R, Flaster E et al. Familial aggregation of Paget’s disease of bone. J Bone Miner Res 1991; 6(5): 495–500. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.5650060511>.
- Cooper C, Dennison E, Schafheutle K et al. Epidemiology of Paget’s disease of bone. Bone 1999; 24(5 Suppl): 3S-5S. Dostupné z DOI: <http://dx.doi.org/10.1016/s8756–3282(99)00023-x>.
- Rendina D, Gennari L, De Filippo G et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21(12): 1828–1835. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.060822>.
- Morales-Piga AA, Bachiller-Corral FJ, Abraira V et al. Is clinical expressiveness of Paget’s disease of bone decreasing? Bone 2002; 30(2): 399–403. Dostupné z DOI: <http://dx.doi.org/10.1016/s8756–3282(01)00674–3>.
- Doyle T, Gunn J, Anderson G et al. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31(5): 616–619. Dostupné z DOI: <http://dx.doi.org/10.1016/s8756–3282(02)00876–1>.
- Tiegs RD, Lohse CM, Wollan PC et al. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27(3): 423–427. Dostupné z DOI: <http://dx.doi.org/10.1016/s8756–3282(00)00333–1>.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45(1–2): 43–48. Dostupné z DOI: <http://dx.doi.org/10.1016/j.clinbiochem.2011.09.026>.
- Siris ES, Ottman R, Flaster E et al. Familial aggregation of Paget’s disease of bone. J Bone Miner Res 1991; 6(5): 495–500. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.5650060511>.
- Seton M, Choi HK, Hansen MF et al. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone – the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18(8): 1519–1524. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.2003.18.8.1519>.
- Merlotti D, Gennari L, Galli B et al. Characteristics and familial aggregation of Paget’s disease of bone in Italy. J Bone Miner Res 2005; 20(8): 1356–1364. Dostupné z DOI: <http://dx.doi.org/10.1359/JBMR.050322>.
- Morales-Piga AA, Rey-Rey JS, Corres-González J et al. Frequency and characteristics of familial aggregation of Paget’s disease of bone. J Bone Miner Res 1995; 10(4): 663–670. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.5650100421>.
- Alonso N, Calero-Paniagua I, Del Pino-Montes J. Clinical and Genetic Advances in Paget’s Disease of Bone: a Review. Clin Rev Bone Miner Metab 2017; 15(1): 37–48. Dostupné z DOI: <http://dx.doi.org/10.1007/s12018–016–9226–0>.
- Chung PY, Beyens G, Boonen S et al. The majority of the genetic risk for Paget’s disease of bone is explained by genetic variants close to the CSF1, OPTN, TM7SF4, and TNFRSF11A genes. Hum Genet 2010; 128(6): 615–626. Dostupné z DOI: <http://dx.doi.org/10.1007/s00439–010–0888–2>.
- Albagha OM, Visconti MR, Alonso N et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet 2010; 42(6): 520–524. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.562>.
- Albagha OM, Wani SE, Visconti MR et al. Genome-wide association identifies three new susceptibility loci for Paget’s disease of bone. Nat Genet 2011; 43(7): 685–689. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.845>.
- Beauregard M, Gagnon E, Guay-Bélanger S et al. Identification of rare genetic variants in novel loci associated with Paget’s disease of bone. Hum Genet 2014; 133(6): 755–768. Dostupné z DOI: <http://dx.doi.org/10.1007/s00439–013–1409-x>.
- Usategui-Martín R, García-Aparicio J, Corral-Gudino L et al. Polymorphisms in autophagy genes are associated with paget disease of bone. PLoS One 2015; 10(6): e0128984. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pone.0128984>.
- Laurin N, Brown JP, Morissette J et al. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70(6):1582–1588. Dostupné z DOI: <http://dx.doi.org/10.1086/340731>.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(Suppl 2): P38-P44. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.06s207>.
- Rea SL, Walsh JP, Ward L et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21(7): 1136–1145. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.060405>.
- Cundy T, Rutland MD, Naot D et al. Evolution of Paget’s disease of bone in adults inheriting SQSTM1 mutations. Clin Endocrinol (Oxf) 2015; 83(3): 315–319. Dostupné z DOI: <http://dx.doi.org/10.1111/cen.12741>.
- Goode A, Long JE, Shaw B et al. Paget disease of bone-associated UBA domain mutations of SQSTM1 exert distinct effects on protein structure and function. Biochim Biophys Acta 2014; 1842(7): 992–1000. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bbadis.2014.03.006>.
- Hiruma Y, Kurihara N, Subler MA et al. A SQSTM1/p62 mutation linked to Paget’s disease increases the osteoclastogenic potential of the bone microenvironment. Hum Mol Genet 2008; 17(23): 3708–3719. Dostupné z DOI: <http://dx.doi.org/10.1093/hmg/ddn266>.
- Kurihara N, Hiruma Y, Zhou H et al. Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest 2007; 117(1):133–142. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI28267>.
- Reddy SV, Kurihara N, Menaa C et al. Osteoclasts formed by measles virus-infected osteoclast precursors from hCD46 transgenic mice express characteristics of pagetic osteoclasts. Endocrinology 2001; 142(7): 2898–2905. Dostupné z DOI: <http://dx.doi.org/10.1210/endo.142.7.8255>.
- Kurihara N, Reddy SV, Menaa C et al. Osteoclasts expressing the measles virus nucleocapsid gene display a pagetic phenotype. J Clin Invest 2000; 105(5):607–614. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI8489>.
- Kurihara N, Hiruma Y, Yamana K et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13(1): 23–34. Dostupné z DOI: <http://dx.doi.org/10.1016/j.cmet.2010.12.002>.
- Teramachi J, Nagata Y, Mohammad K et al. Measles virus nucleocapsid protein increases osteoblast differentiation in Paget’s disease. J Clin Invest 2016; 126(3): 1012–1022. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI82012>.
- Mills BG, Singer FR, Weiner LP et al. Cell cultures from bone affected by Paget’s disease. Arthritis Rheum 1980; 23(10): 1115–1120. Dostupné z DOI: <http://dx.doi.org/10.1002/art.1780231007>.
- Rebel A, Basle M, Pouplard A et al. Towards a viral etiology for Paget’s disease of bone. Metab Bone Dis Relat Res 1981; 3(4–5): 235–238. Dostupné z DOI: <http://dx.doi.org/10.1016/0221–8747(81)90038–2>.
- Singer FR. Paget’s disease of bone-genetic and environmental factors. Nat Rev Endocrinol 2015; 11(11): 662–671. Dostupné z DOI: <http://dx.doi.org/10.1038/nrendo.2015.138>
- Devogelaer JP, Bergmann P, Body JJ et al. Management of patients with Paget’s disease: a consensus document of the Belgian Bone Club. Osteoporos Int 2008; 19(8): 1109–1017. Dostupné z DOI: <http://dx.doi.org/10.1007/s00198–008–0629–8>.
- Siris E, Roodman GD. Paget Disease Section X. In: Favus MJ (ed). Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. ASBMR: Washington (DC) 2003. ISBN 978–0974478203.
- Dabbs TR, Skjodt K. Prevalence of angioid streaks and other ocular complications of Paget’s disease of bone. Br J Ophthalmol 1990; 74(10): 579–582. Dostupné z DOI: <http://dx.doi.org/10.1136/bjo.74.10.579>.
- Yost JH, Spencer-Green G, Krant JD. Vascular steal mimicking compression myelopathy in Paget’s disease of bone: rapid reversal with calcitonin and systemic steroids. J Rheumatol 1993; 20(6): 1064–1065.
- Whyte MP. Clinical practice. Paget’s disease of bone. N Engl J Med 2006; 355(6): 593–600. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMcp060278>.
- Redden JF, Dixon J, Vennart W et al. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5(2): 103–106. Dostupné z DOI: <http://dx.doi.org/10.1007/BF00267839>.
- Wermers RA, Tiegs RD, Atkinson EJ et al. Morbidity and mortality associated with Paget’s disease of bone: a population-based study. J Bone Miner Res. 2008; 23(6): 819–825. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.080215>.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; Suppl 2: P58-P63. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.06s211>.
- Ralston SH, Langston AL, Reid IR. Pathogenesis and management of Paget’s disease of bone. Lancet 2008; 372(9633): 155–163. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(08)61035–1>.
- Rendina D, Mossetti G, Soscia E et al. Giant cell tumor and Paget’s disease of bone in one family: geographic clustering. Clin Orthop Relat Res 2004; (421): 218–224. Dostupné z DOI: <http://dx.doi.org/10.1097/00000118702.46373.e3>.
- Rendina D, De Filippo G, Ralston SH et al. Clinical characteristics and evolution of giant cell tumor occurring in Paget’s disease of bone. J Bone Miner Res 2015; 30(2): 257–263. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.2349>.
- Alvarez L, Guañabens N, Peris P et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29(5): 447–452. Dostupné z DOI: <http://dx.doi.org/10.1016/s8756–3282(01)00592–0>.
- Reid IR, Davidson JS, Wattie D et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35(1): 224–230. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bone.2004.03.023>.
- Peris P, Alvarez L, Vidal S et al. Biochemical response to bisphosphonate therapy in pagetic patients with skull involvement. Calcif Tissue Int 2006; 79(1):22–26. Dostupné z DOI: <http://dx.doi.org/10.1007/s00223–005–0247–9>.
- Shankar S, Hosking DJ. Biochemical assessment of Paget’s disease of bone. J Bone Miner Res 2006; 21(Suppl 2): P22-P27. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.06s204>.
- Seton M. Paget’s disease of bone. In: Hochberg MC, Silman AJ, Smolen JS et al (eds). Rheumatology. 4th ed. Mosby (Elsevier): Philadelphia 2007. ISBN 978–0323044301.
- Griffiths HJ. Radiology of Paget’s disease. Curr Opin Radiol 1992; 4(6):124–128.
- Singer FR, Bone HG, Hosking DJ et al. Paget’s disease of bone: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99(12): 4408–4422. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2014–2910>.
- Ralston SH, Corral-Gudino L, Cooper C et al. Diagnosis and Management of Paget’s Disease of Bone in Adults: A Clinical Guideline. J Bone Miner Res 2019; 34(4): 579–604. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.3657>.
- Whyte MP. Clinical practice. Paget’s disease of bone. N Engl J Med 2006; 355(6): 593–600. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMcp060278>.
- Sundaram M, Khanna G, El-Khoury GY. T1-weighted MR imaging for distinguishing large osteolysis of Paget’s disease from sarcomatous degeneration. Skeletal Radiol 2001; 30(7): 378–383. Dostupné z DOI: <http://dx.doi.org/10.1007/s002560100360>.
- Sundaram M. Imaging of Paget’s disease and fibrous dysplasia of bone. J Bone Miner Res 2006; 21(Suppl 2): P28-P30. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.06s205>
- Spieth ME, Kasner DL, Manor WF. Positron emission tomography and Paget disease: hot is not necessarily malignant. Clin Nucl Med 2003; 28(9): 773–774. Dostupné z DOI: <http://dx.doi.org/10.1097/01.rlu.0000082671.73091.db>.
- Singer FR, Bone HG 3rd, Hosking DJ et al. Paget’s disease of bone: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014; 99(12): 4408–4422. Dostupné z DOI: <http://dx.doi.org/10.1210/jc.2014–2910>.
- Reid IR, Nicholson GC, Weinstein RS et al. Biochemical and radiologic improvement in Paget’s disease of bone treated with alendronate: a randomized, placebo-controlled trial. Am J Med 1996; 101(4): 341–348. Dostupné z DOI: <http://dx.doi.org/10.1016/s0002–9343(96)00227–6>.
- Siris E, Weinstein RS, Altman R et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81(3): 961–967. Dostupné z DOI: <http://dx.doi.org/10.1210/jcem.81.3.8772558>.
- Walsh JP, Ward LC, Stewart GO et al. A randomized clinical trial comparing oral alendronate and intravenous pamidronate for the treatment of Paget’s disease of bone. Bone 2004; 34(4): 747–754. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bone.2003.12.011>.
- Brown JP, Chines AA, Myers WR et al. Improvement of pagetic bone lesions with risedronate treatment: a radiologic study. Bone 2000; 26(3): 263–267. Dostupné z DOI: <http://dx.doi.org/10.1016/s8756–3282(99)00271–9>.
- Ralston SH, Corral-Gudino L, Cooper C et al. Diagnosis and management of Paget’s disease of bone in adults: a clinical guideline. J Bone Miner Res 2019; 34(4): 579–604. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.3657>.
- Reid IR, Miller P, Lyles K et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353(9): 898–908. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa044241>.
- Reid IR, Lyles K, Su G et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26(9): 2261–2270. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.438>.
- Cundy T, Maslowski K, Grey A et al. Durability of Response to Zoledronate Treatment and Competing Mortality in Paget’s Disease of Bone. J Bone Miner Res 2017; 32(4):753–756. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.3029>
- Merlotti D, Gennari L, Martini G et al. Comparison of different intravenous bisphosphonate regimens for Paget’s disease of bone. J Bone Miner Res 2007; 22(10): 1510–1517. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.070704>.
- Hosking D, Lyles K, Brown JP et al. Long-term control of bone turnover in Paget’s disease with zoledronic acid and risedronate. J Bone Miner Res 2007; 22(1): 142–148. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.061001.22:142>.
- Miller PD, Roux C, Boonen S et al. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20(12): 2105–2115. Dostupné z DOI: <http://dx.doi.org/10.1359/JBMR.050817>.
- Jamal SA, Bauer DC, Ensrud KE et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22(4): 503–508. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.070112>.
- Miller PD. The kidney and bisphosphonates. Bone 2011; 49(1): 77–81. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bone.2010.12.024>.
- Reid IR, Sharma S, Kalluru R et al. Treatment of Paget’s Disease of Bone with Denosumab: Case Report and Literature Review. Calcif Tissue Int 2016; 99(3): 322–325. Dostupné z DOI: <http://dx.doi.org/10.1007/s00223–016–0150–6>.
- Huynh AL, Baker ST, Stewardson AJ et al. Denosumab-associated hypocalcaemia: incidence, severity and patient characteristics in a tertiary hospital setting. Pharmacoepidemiol Drug Saf 2016; 25(11): 1274–1278. Dostupné z DOI: <http://dx.doi.org/10.1002/pds.4045>.
- Jalleh R, Basu G, Le Leu R et al. Denosumab-Induced Severe Hypocalcaemia in Chronic Kidney Disease. Case Rep Nephrol 2018; 2018: 7384763. Dostupné z DOI: <http://dx.doi.org/10.1155/2018/7384763>.
- Parvizi J, Schall DM, Lewallen DG et al. Outcome of uncemented hip arthroplasty components in patients with Paget’s disease. Clin Orthop Relat Res 2002; (403): 127–134. Dostupné z DOI: <http://dx.doi.org/10.1097/00003086–200210000–00020>.
- Hernandez NM, Vakharia RM, Mont MA et al. Paget’s disease in primary total hip arthroplasty is associated with greater in-hospital lengths of stay, costs, and complications. J Arthroplasty 2020; S0883–5403(20)30896–2. Dostupné z DOI: <http://dx.doi.org/10.1016/j.arth.2020.08.017>.
- Kaplan FS. Surgical management of Paget’s disease. J Bone Miner Res 1999; 14(Suppl 2): 34–38. Dostupné z DOI: <http://dx.doi.org/10.1002/jbmr.5650140208>.
- Wegrzyn J, Pibarot V, Chapurlat R et al. Cementless total hip arthroplasty in Paget’s disease of bone: a retrospective review. Int Orthop 2010; 34(8): 1103–1109. Dostupné z DOI: <http://dx.doi.org/10.1007/s00264–009–0853–7>.
- Whitson HE, Lobaugh B, Lyles KW. Severe hypocalcemia following bisphosphonate treatment in a patient with Paget’s disease of bone. Bone 2006; 39(4): 954–958. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bone.2006.04.032>.
- Alvarez L, Peris P, Guañabens N et al. Long-term biochemical response after bisphosphonate therapy in Paget’s disease of bone. Proposed intervals for monitoring treatment. Rheumatology (Oxford) 2004; 43(7): 869–874. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/keh185>.
- Reid IR, Maslowski K. Long-Term Bone Scintigraphy Results After Intravenous Zoledronate in Paget’s Disease of Bone. Calcif Tissue Int 2017; 101(1): 43–49. Dostupné z DOI: <http://dx.doi.org/10.1007/s00223–017–0261–8>.
- Siris ES, Feldman F. Natural history of untreated Paget’s disease of the tibia. J Bone Miner Res 1997; 12(4): 691–692. Dostupné z DOI: <http://dx.doi.org/10.1359/jbmr.1997.12.4.691>.
Štítky
Biochemie Dětská gynekologie Dětská radiologie Dětská revmatologie Endokrinologie Gynekologie a porodnictví Interní lékařství Ortopedie Praktické lékařství pro dospělé Radiodiagnostika Rehabilitační a fyzikální medicína Revmatologie Traumatologie OsteologieČlánek vyšel v časopise
Clinical Osteology

2020 Číslo 4
Nejčtenější v tomto čísle
- Diagnostika a léčba Pagetovy kostní choroby: aktualizace 2020
- Nutriční (vitamin D deficitní) rachitis – opomíjená diagnóza? Kazuistika
- Může být FRAX přínosnější? Editorial
- Kost a imunitní systém – osteoimunologie